Introduction à la modélisation moléculaire. TD1: visualisation.
Date: 14 fevrier 2019
E. Dumont
Nous allons utiliser et nous familiariser avec le logiciel Avogadro, qui est gratuit et facilement installable. Nous allons nous familiariser avec la construction de molécules, et évaluer les énergies en utilisant des champs de forces.
A) Construction de molécules simples
- Lancer Avogadro. Une fenetre apparait avec un menu don’t nous allons explorer les fonctionnalités ensemble.
- Construire à la souris la molécule de cyclobutane. On pensera à verifier qu’elle comporte le bon nombre d’hydrogènes. Selon la manière dont vous procéder, la molécule est plus ou moins réaliste...
- Pour obtenir une structure plus réaliste minimisant l’énergie de cette molécule, on peut employer différentes méthodes de modélisation:
- La première est purement géométrique: elle correspond dans le menu à Optimize Geometry dans l’onglet Extensions. Relever les distances après avoir réalisé cette optimization de géométrie. (Indication: vous pouvez mesurer en pointant les atomes avec la souris, mais également retrouver toutes les informations structurales dans le Menu View, Properties.
- Une méthode de mécanique moléculaire, associé à un jeu de paramètres regroupés dans un champ de force. elle correspond dans le menu à
Optimize Geometry dans l’onglet Extensions. Lister les champs de forces disponibles dans Avogadro et reporter l'énergie.
- Une méthode de chimie quantique avec un hamiltonien. On verra dans la suite de ce cours qu’on dispose de 3 familles de méthodes: les méthodes semi-empiriques, la méthode Hartree-Fock et les méthodes dites de la théorie de la fonctionnelle de la densité (DFT). Elles ne sont pas implémentées dans Avogadro, et il faut alors coupler ce logiciel de visualisation à un programme de chimie quantique: Orca.
- Toutes les données expérimentales pour ce type de “petite” molécules sont mises à disposition sur le serveur Chemistry Webbook de la NIST (National Institute for Standards and Technology). Quelles sont les informations disponibles pour la molécule de cyclobutane ?
On téléchargera la structure proposée au format .mol et on l’ouvrira dans Avogadro (File / Open). Quelles sont les distances interatomiques entre les atomes de carbone ?
B) Une réaction importante en biochimie
Il s'avère parfois délicat voire dangereux de construire “à la main” une molécule. C’est d’autant plus vrai que la molécule présente différents conformères, et qu’elle est cyclique ou de taille importante. Pour les molécules biologiques cela devient impossible.
La Protein DataBank permet de charger en ligne des structures obtenues expérimentalement (principalement par cristallographie RX, mais aussi par RMN ou cryo EM). Nous allons nous intéresser à la formation de cyclobutanes dans l'ADN. Un schéma pour leur formation par voie photochimie est donnée ci-dessous:
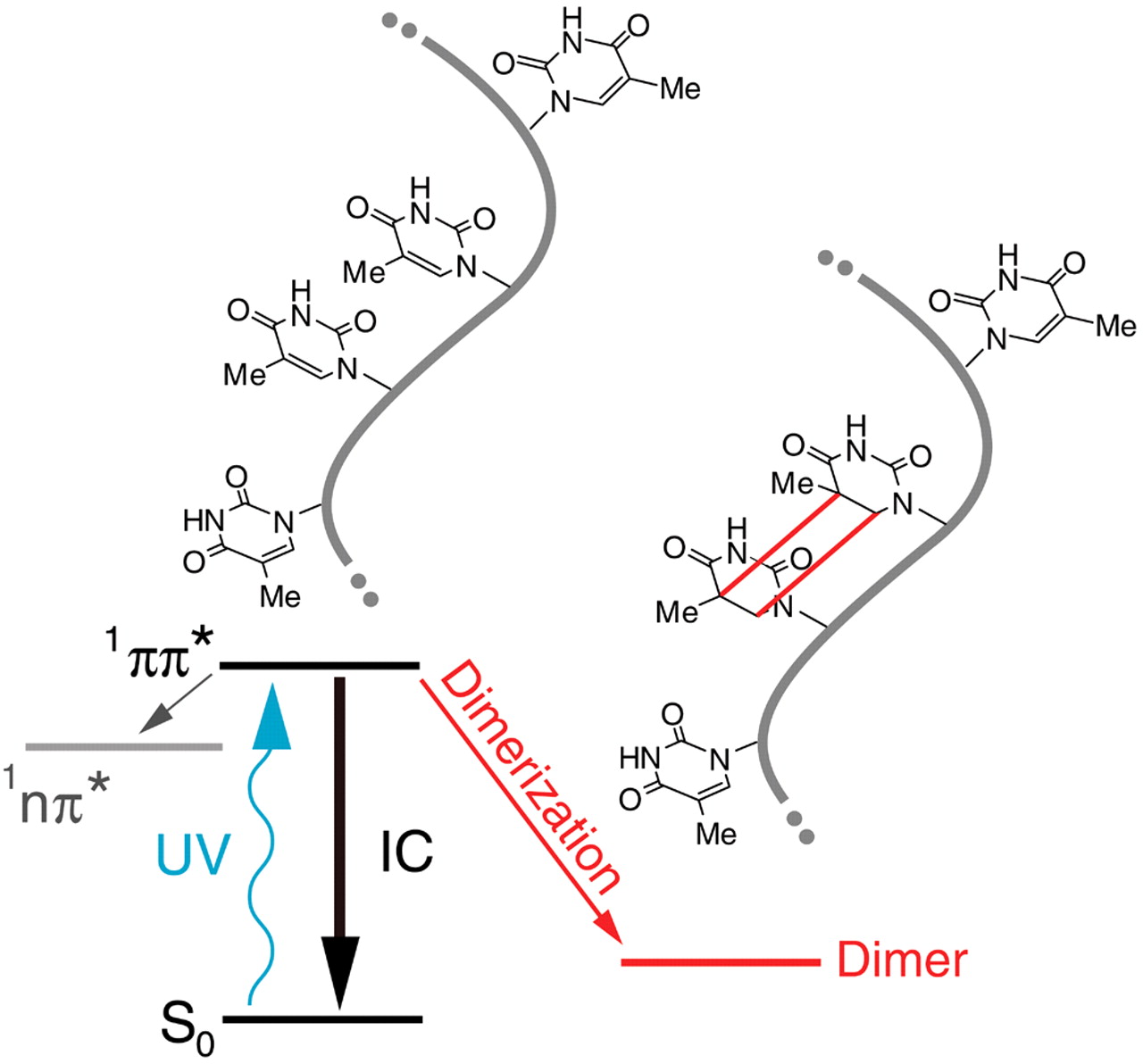
- Télécharger la structure de numéro 1QL5 (lien) et l’ouvrir avec Avogadro. Localiser le cyclobutane et mesurer les distances C-C caractéristiques. Analyser ce résultat.
- Quelle réaction de dimérisation peut donner lieu à la formation de dérivés du cyclobutane ?
Evaluer la faisabilité thermodynamique de cette réaction.
Pour pouvoir étudier les aspects cinétiques de cette réaction, il faudra une description du profil réactionnel, ce qui appelle des méthodes de chimie quantique notamment pour estimer l'énergie de l'état de transition.
C) Jouons avec des fragments
Puisqu'il est risqué de construire "à la main" des molécules dès qu'elles dépassent une certaine taille, Avogadro dispose de librairies de géométries déjà implémentées. Elles se trouvent dans l'onglet Build, puis Insert.
- Une possibilité est de construire à l'aide de fragments plutot que de manipuler des atomes un à un.
Construire une séquence d'ADN en utilisant le module dédié.
- Explorer, dans le temps restant de cette séance, les molécules proposées dans la bibliothéque nommée "Fragments".
Last update: Feb. 2019
|