TP Modélisation: application à la chimie supramoléculaire.
Date: Sept. 2021
E. Dumont, S. Steinmann, P. Colinet
Cette séance se centre sur une problématique de chimie supramoléculaire, où la recherche de cage fonctionnellles est guidée par les outils expérimentaux usuels (cristallographie, RMN, ITC) mais met également en jeu un éclairage par la chimie computationnelle. Le système est emprunté à cette publication: J. Org. Chem. 2016:654–661 lien.
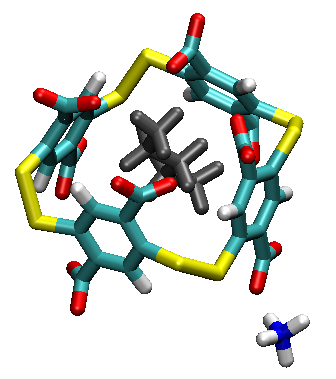
Sur ce système, nous allons pouvoir illustrer les points suivants:
Objectif: acquérir plus d'autonomie sur des premiers calculs de chimie quantique, comprendre les différences entre HF et
DFT, avoir une notion de l'impact d'un choix de base.
3. Hartree-Fock en pratique: quelles informations dans le fichier de sortie ?
Pour un premier calcul quantique ab initio, nous allons reprendre le fichier d'entrée (input) et lancer un calcul d'énergie
au niveau HF/STO-3G. Pour cela, il suffit de remplacer le mot-clé am1 par hf/sto-3g et ne pas demander d'optimisation de géométrie. En effet, pour un calcul
quantique on doit se munir d'une base d'orbitales alors qu'avec une méthode semi-empirique elle est choisie pour vous implicitement.
On demande de lancer le calcul et d'analyser le fichier de sortie en se guidant des questions suivantes.
On rappelle l'input car il faut introduire des mots-clés spécifiques pour que les orbitales et toutes les contributions énergétiques qui
vont nous intéresser soient écrites:
%chk=geometrie-xray-hf.chk
%mem=2000Mb
#p hf/sto-3g gfinput iop(6/7=3) Extralink=608
Calcul energie HF/STO-3G
0 1
[xyz]
00
A vous de jouer !
Pour cette partie, on pourra s'aider du fichier de sortie Gaussian commenté ici.
- Combien d'atomes et d'électrons sont considérés ?
- Trouver les constantes rotationnelles pour la géométrie de départ.
- Quel est le nombre de fonctions de base (NBasis) ? Retrouvez à quoi correspond l'acronyme STO-3G en vous aidant de votre cours.
- Quelle est l'énergie totale HF pour la géométrie de départ ?
- On va s'intéresser aux différentes contributions pour ce terme énergétique total, qui sont données en amont dans le fichier de sortie.
Identifier les termes ET, EV, EJ, EK, et Enuc, en commentant le signe et la grandeur énergétique.
Le terme EV ne s'affiche pas avec la compilation par défaut mais on pourra la retrouver à l'aide du terme ENTV. Pourquoi a-t-on Ec=0 ?
- Combien d'OMs figurent dans le fichier de sortie ? Combien d'orbitales de coeur identifiez-vous ? Quelles sont les énergies de la HOMO et de
la LUMO ?
- Lancer sur la meme structure une optimisation de géométrie. Vérifier que la convergence de l'optimisation a bien été atteinte. Quelle distance est prévue pour les ponts disulfures ? On reportera aussi les angles dièdres.
- (Optionel). On pourra recalculer les écarts entre les différents conformères pour comparer AM1 avec HF/STO-3G, et avec le niveau de
théorie de la publication originale.
4. Les bases d'OAs
Dès lors que nous allons utiliser des calculs quantiques ab initio et des bases de qualité standard et non la base minimale STO-3G, le temps de calcul va devenir plus significatif. Dans le reste de cette séance, on va surtout s'intéresser à l'analyse des fichiers de sortie qui vous sont fournis. Cela nous permettra de balayer les grandeurs et informations accessibles par un calcul quantique.
PPPP-opt-hf.log, PPMM-opt-hf.log, MPMP-opt-hf.log et
MMPP-opt-hf.log.
- Quel est le nombre de fonctions de base (NBasis) ?
- Quelle est l'énergie totale de la molécule au niveau de calcul HF/6-31G* ? Se compare-t-elle à l'énergie trouvée au niveau de calcul HF/STO-3G ?
- De combien le temps de calcul a-t-il été augmenté ? (Information donnée en dernière ligne.)
5. Un premier calcul au niveau DFT: quelles informations dans le fichier de sortie ?
Pour améliorer la précision du calcul et pouvoir etre prédictif,
on recourt le plus souvent à un calcul basé sur la théorie de la fonctionnelle de la densité (DFT).
Pour ce calcul, on doit se murir d'une fonctionnelle, c'est-à-dire d'une fonction mathématique qui fournit une énergie électronique à partir de la densité électronique. Ici, on prendra la fonctionnelle B3LYP (voir cours pour plus d'explication). Contrairement aux calculs faisant intervenir un hamiltonien semi-empirique, on a besoin de se munir d'une base d'orbitales atomiques, ici 6-31G* (ou de manière équivalente 6-31G(d)).
Le fichier de sortie est conséquent sur les calculs de structure atomiques. Nous verrons en cours quelles sont les différentes étapes:
changement d'axes pour la molécule, generation d'une fonction d'onde d'essai, etc...
A vous de jouer !
- On vous donne le résultat de l'optimisation de géométrie pour les 4 conformères PPPP-opt-dft.log, MMPP-opt-dft.log, MPMP-opt-dft.log et MMPP-opt-dft.log.
, à partir d'un nouveau fichier d'entrée avec pour mots-clés: # b3lyp/6-31G* empiricaldisp=gd3bj opt.
Analyser la différence de géométrie entre la structure cristallographique, la géométrie obtenue après optimisation au niveau AM1 et au niveau
de calcul B3LYP/6-31G*.
- Quelle est l'énergie totale finale de la molécule ? (ligne "SCF Done")
- Cette énergie totale se compose de cinq termes que l'on présentera en cours.
Que vaut l'énergie de répulsion nucléaire ? l'énergie cinétique, potentielle et inter-électronique ? On identifiera également les contributions d'échange et de corrélation. On demande de traiter cette question sur la meme structure que celle utilisée pour le calcul HF MPMP-opt-dft.log.
- Que vaut l'énergie de dispersion, qui est incluse comme correction de cette fonctionnelle ?
- Combien de cycles DFT ont été nécessaires pour l'optimisation de géométrie ?
- Il est possible une fois cette géométrie optimisée obtenue de conduire un calcul de fréquences, notamment pour vérifier que l'on a bien obteni un minimum. Cela correspond au calcul du hessien (dérivées secondes par rapport à l'énergie). En reprenant la géométrie optimisée DFT, on lance un calcul de fréquences par le mot-clé freq. Le fichier de sortie est donné: opt-freq.log.
Quelles fréquences ressortent et à quels modes de vibration correspondent-elles ? Pourquoi certaines ont une intensité nulle ? On comparera le spectre IR prédit à celui obtenu au niveau de calcul AM1.
- Evaluer les énergies relatives des 4 conformères. Que constatez-vous ? On comparera au niveau de calcul MP2 de l'article de reference.
- La géométrie du cyclophane s'avère assez différente par rapport à sa formule semi-développée, qui notamment ne reflète pas de l'équilibre entre plusieurs conformères. Il est possible de demander à Gaussian de sortir la fonction d'onde (wavefunction, au format .wfn) et de l'analyser en termes
d'interactions non covalentes. Cela peut donner un début d'explication pour le conformère le plus stable.
Aparté: on peut extraite la fonction d'onde pour visualiser qualitativement les interactions de dispersion stabiliser la cage meme si les ponts disulfures imposent une géométrie non empilée. Cet effet est général en chimie supramoléculaire.
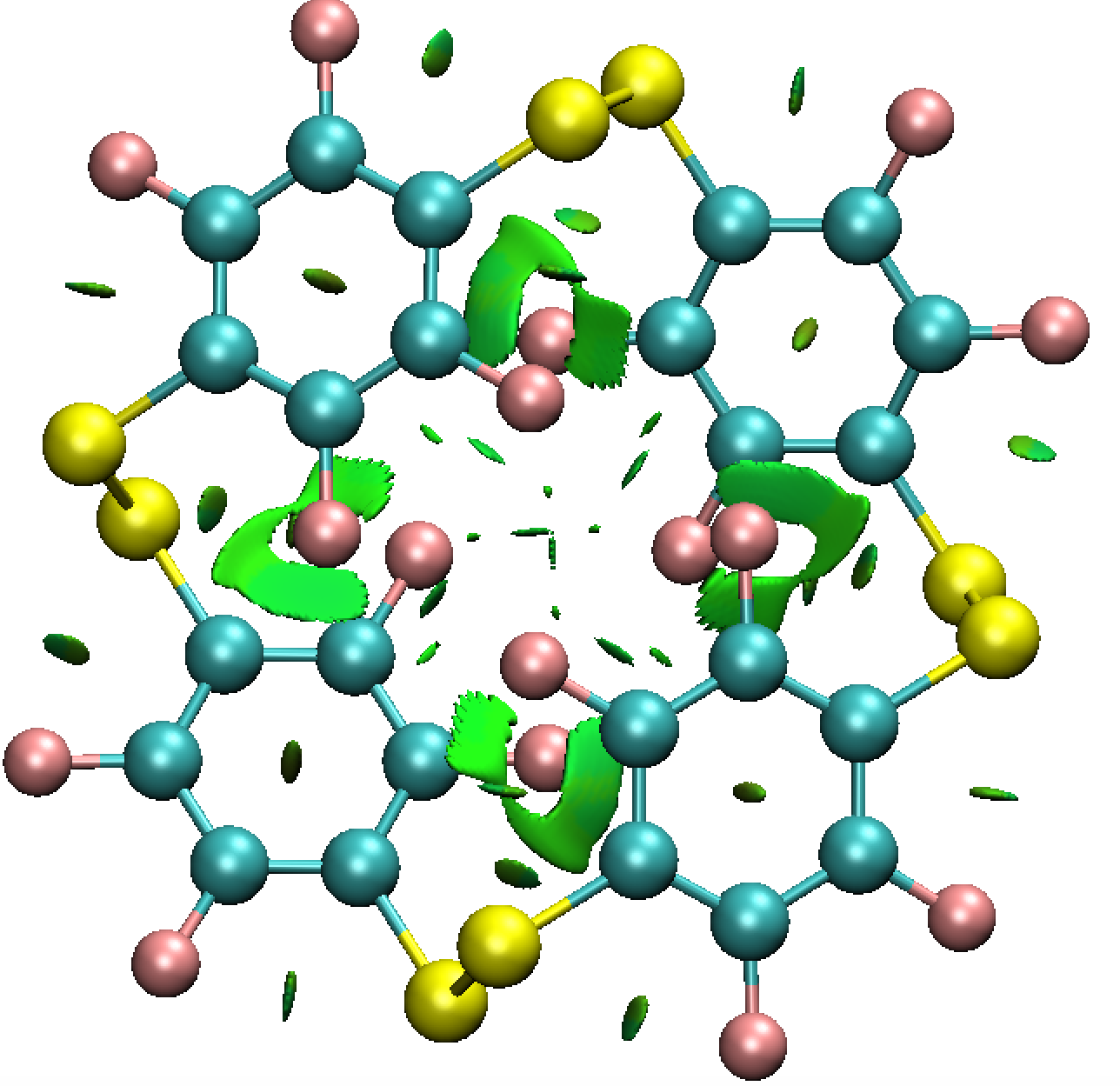
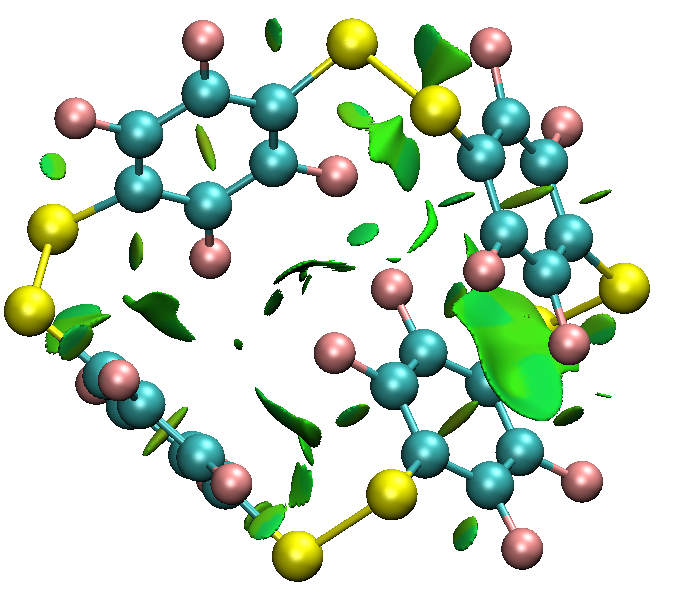
6. Association hote-invité : signature par spectroscopie UV/Vis et attribution des transitions électronique
Ces cages sont désignées pour capter des anions halogènures, selon des interaction π--anion. Les structures cristallographiques ne sont disponibles que pour la cage. Pour proposer une structure plausible, on peut alors s'appuyer sur des calculs pour mieux caractériser
cette interaction.
- Pour l'iode, il convient d'utiliser une base de type 6-31+G**. Expliquer pourquoi ? (en vous aidant du cours !)
- Pour la cage MPMP avec ion iodure, on donne le fichier de sortir correspondant à une optimisation de géométrie au niveau de calcul DFT/B3LYP-D3BJ: cage-with-i.log. Estimer l'énergie d'association. On aura besoin d'un (court !) calcul annexe que l'on précisera.
- A l'aide d'une information facilement accessible, évaluer le transfert de charge entre molécule invitée et la cage.
- On donne pour les ions fluorures et chlorures les résultats d'une optimisation de géométrie:
cage-with-f.log, cage-with-cl.log et cage-with-br.log
Commenter les énergies d'association relatives et le transfert de charges. Pourquoi l'énergie de l'anion bromure est-elle plus basse que
celle de l'anion fluor ?
- Cette association est délicate à caractériser expérimentalement: on peut mesurer la constante d'association par ITC, et également mettre en évidence la présence d'une bande à transfert de charge en spectroscopie UV-Vis. On donne les calculs TDDFT qui permetente de situer les transitions verticales : cage-avec-i-tddft.log et cage-sans-i-tddft.log. Identifier la
nature des orbitales en jeu et comparer avec le spectre reporté expérimentalement. On donne le script suivant: spectrum.py.
Pour avoir une meilleure énergétique du système plus en accord avec les ITC, il faudrait procéder par dynamique moléculaire. Ici le systeme est assez rigide pour qu'on obtienne de bons résultats par la DFT ("cage moins rigide"). De cette manière, on peut etre quantitatif et non seulement reproduire la tendance le long des ions halogènures. On peut également aller chercher la signature RMN, RPE, de dichroisme circulaire, Raman ..
Last update: Sept. 2019
|