TP L3 – Séance 2
Isomères et conformères
Date: Avril 2019
C. Michel, E. Dumont, P. Clabaut
Les compétences à acquérir :
- Déterminer quelle est la meilleure méthode pour des données thermodynamiques;
- Recherche conformationnelle simple
- Optimisation de géométrie contrainte
- Notion de modèles
- Travailler à plusieurs
La session se divise en 2 parties
1.1 Grandeurs thermodynamiques
- Lister les isomères de constitution du cyclobutane.
- Optimiser le cyclobutane au niveau AM1 et déterminer son enthalpie libre, entropie et enthalpie libre (cf. TP précédent). Quelles sont les approximations faites pour évaluer ces grandeurs?
- Faites de même avec les isomères choisis au même niveau de calcul. ( Pensez à vous partager le travail ! ) .
- Calculer l’enthalpie, d’isomérisation duc cyclobutane vers chacun des isomères choisis. Comparer avec les données disponibles dans les bases de données en ligne. Les résultats vous paraissent-ils cohérents ?
- Utilisez maintenant la méthode RHF pour évaluer ces différences d’enthalpie. Prêtez attention à la durée du calcul. Conclure quand à la meilleure méthode pour évaluer cette isomérisation et à ses limites.
- Nous allons maintenant chercher à déterminer l’énergie de tension de cycle du cylobutane. Pour cela, il faut déterminer l’énergie de réaction de formation du cycle en gardant toute chose égale par ailleurs (hybridation des carbones, nombre exacte de liaisons de chaque type,… ). Ce genre de réaction est dite « homodesmique ». Dans le cas du cyclobutane, cela correspond à la réaction suivante:
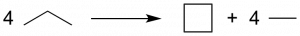
- Déterminer la tension de cycle pour le cyclobutane (méthode de calcul au choix).
- Déterminer la tension de cycle pour le cyclopropane, le cyclopentane et le cyclohexane.
- Commenter les résultats.
1.2 Structure électronique et énergie d’ionisation
- Visualiser les orbitales moléculaires du cyclobutane avec Avogadro (voir séance 1 ou manuel). Identifier-les.
- Rappeler le théorème de Koopman.
- Déterminer l’énergie du cation C4H8+ correspondants en gardant bien la géométrie fixée (simple point) en prenant garde à la charge et à la multiplicité de spin. Réfléchissez à la signification de l’ionisation « verticale ». Attention ! Comme il s’agit ici d’une molécule ayant un spin différent de 1, la méthode RHF doit être substitué par la méthode UHF (Unrestricted). Si vous optez pour cette méthode, pensez à vérifier que les deux (UHF et RHF) vous donnent la même énergie pour la géométrie optimisée du cyclobutane.
- Déterminer la géométrie optimale de ces cations. Comment la géométrie évolue? Comment le potentiel de ionisation évolue-t-il si la ionisation n’est pas verticale? Comment la structure électronique évolue-t-elle? Comparer vos résultats aux résultats expérimentaux que vous trouverez dans Chemistry Webbook
Cette partie cherche à comprendre l’origine de la chiralité du binaphtol puis analyser les interactions permettant son dédoublement en utilisant le chlorure de N-benzylcinchonidinium.
Le binaphtol étant une ‘grosse’ molécule, nous l’aborderons par étapes. Nous commençons par étudier la formation de liaisons hydrogène dans des diols terminaux.
- Optimiser le 1,2-ethanediol, le 1,3-propanediol et le 1,4-butanediol avec et sans liaison hydrogène en choisissant votre niveau de calcul. Quelle est la stabilité fournie par la liaison hydrogène?
- Pour obtenir l’énergie en fonction de l’angle dièdre de rotation, nous allons réaliser une série d’optimisation contraintes en utilisant le mot-clé moderedundant en option du mot-clé opt.
Plus d’information ici ou dans le manuel. Un exemple suit pour un échantillonnage de 36 points espacés d’un angle dièdre de 10° pour le 1,2-ethanediol.#n PM3 Opt=modredundant Scan 0 1 C -1.05977 0.44370 -0.51666 C 0.46533 0.40419 -0.42841 H -1.50404 0.72782 0.46481 H -1.43768 -0.57106 -0.76256 O -1.47403 1.32722 -1.52351 H 0.78967 -0.34237 0.33164 O 0.97864 1.67041 -0.11480 H 0.88509 0.08160 -1.40478 H -1.46854 2.23712 -1.12648 H 0.82582 1.80626 0.85677 5 1 2 7 S 36 10.0
- Quel diol choisissez-vous comme modèle simple du binaphtol (sans cycle aromatique)? Déterminer l’énergie en fonction d’un angle dièdre bien choisi. Pour cela, utilisez le script python fourni en fin de TP afin de lire le fichier .log et extraire les géométries des points optimisés ainsi que les énergies correspondantes. Vous pouvez également construire un film avec ces points en utilisant
cat *step* > movie.xyz
. Il sera visualisable avec Avogadro. Comparer avec les diols vus précédemment.
Maintenant, nous allons étudier le rôle de la partie aromatique du binaphtol.
- Construire le bi-phényl en utilisant des fragments et optimiser-le. Est-il plan? Pourquoi? Déterminer l’angle dièdre autour de la liaison simple. Déterminer le profil d’énergie en fonction autour de cet angle. Comparer avec des données de la littérature.
- Construire la configuration R du binaphtol en utilisant son numéro CAS et avec la fonction import/Fetch by chemical name sur Avogadro. Optimiser la structure. Déterminer l’angle dièdre autour de la liaison simple. Déterminer le profil d’énergie en fonction de cet angle jusqu’à la configuration S. Conclure quand à l’origine de la chiralité en comparant l’ensemble des résultats obtenus.
Une fois le profil énergétique du binapthol seul obtenu, on va s’intéresser à l’effet du chlorure de N-benzylcinchonidinium sur ce profil, afin d’expliquer son rôle d’agent de dédoublement (structure de l’agent de dédoublement avec la configuration R du binapthol).
- Optimiser la géométrie de l’agent de dédoublement seul.
- Construire puis optimiser plusieurs géométries de complexes d’interaction au niveau MM entre l’agent et le binapthol en conformation R ou S. Quelle est l’enthalpie libre de complexation pour le R et pour le S au niveau MM choisi? au niveau AM1? Est-ce concluant?
- Si ce n’est pas concluant, cela signifie que (i) la recherche conformationnelle n’a pas permis de trouver le minimum global (ii) le niveau de calcul ne permet pas de décrire toutes les interactions. En particulier, AM1 ne capture peut être pas correctement les inteactions faibles. Comment vérifier? Quelle alternative?.
Script:
#!/usr/bin/python
from sys import argv
import numpy as np
import matplotlib.pyplot as plt
periodic_table = {1:'H', 2:'He',
3:'Li', 4:'Be', 5:'B', 6:'C', 7:'N', 8:'O', 9:'F', 10:'Ne',
11:'Na', 12:'Mg', 13:'Al', 14:'Si', 15:'P', 16:'S', 17:'Cl', 18:'Ar',
19:'K', 20:'Ca', 21:'Sc', 22:'Ti', 23:'V', 24:'Cr', 25:'Mn', 26:'Fe', 27:'Co', 28:'Ni', 29:'Cu', 30:'Zn', 31:'Ga', 32:'Ge', 33:'As', 34:'Se', 35:'Br', 36:'Kr',
37:'Rb', 38:'Sr', 39:'Y', 40:'Zr', 41:'Nb', 42:'Mo', 43:'Tc', 44:'Ru', 45:'Rh', 46:'Pd', 47:'Ag', 48:'Cd', 49:'In', 50:'Sn', 51:'Sb', 52:'Te', 53:'I', 54:'Xe',
55:'Cs', 56:'Ba', 57:'La',
58:'Ce', 59:'Pr', 60:'Nd', 61:'Pm', 62:'Sm', 63:'Eu', 64:'Gd', 65:'Tb', 66:'Dy', 67:'Ho', 68:'Er', 69:'Tm', 70:'Yb', 71:'Lu',
72:'Hf', 73:'Ta', 74:'W', 75:'Re', 76:'Os', 77:'Ir', 78:'Pt', 79:'Au', 80:'Hg', 81:'Tl', 82:'Pb', 83:'Bi', 84:'Po', 85:'At', 86:'Rn',
87:'Fr', 88:'Ra', 89:'Ac',
90:'Th', 91:'Pa', 92:'U', 93:'Np', 94:'Pu', 95:'Am', 96:'Cm', 97:'Bk', 98:'Cf', 99:'Es', 100:'Fm', 101:'Md', 102:'No', 103:'Lr',
104:'Rf', 105:'Db', 106:'Sg', 107:'Bh', 108:'Hs', 109:'Mt', 110:'Ds', 111:'Rg', 112:'Cn', 113:'Nh', 114:'Fl', 115:'Mc', 116:'Lv', 117:'Ts', 118:'Og'}
input_file = argv[1]
if input_file.endswith(".log"):
input_log = open(input_file, 'r')
basename = input_file.split('.')[0]
log_lines = input_log.readlines()
input_log.close()
# Starting atom variables empty to avoid 'NameError'
scan_param = '0.0'
mod_atom_sym1 = '0.0'
mod_atom_sym2 = '0.0'
mod_atom_sym3 = '0.0'
mod_atom_sym4 = '0.0'
atom1 = '0.0'
atom2 = '0.0'
atom3 = '0.0'
atom4 = '0.0'
coord_data = [ ]
coord_data_LoL = [ ]
scf_data = [ ]
# Flags start 'off'.
mod_flag = 0
xyz_flag = 0
dash_flag = 0
for line in log_lines:
if 'ModRedundant' in line:
mod_flag = 1
if mod_flag == 1 and 'ModRedundant' not in line:
mod_info = line.split()
mod_flag = 0
if len(mod_info) == 6:
scan_param = 'distance'
atom1 = int(mod_info[1])
atom2 = int(mod_info[2])
elif len(mod_info) == 7:
scan_param = 'angle'
atom1 = int(mod_info[1])
atom2 = int(mod_info[2])
atom3 = int(mod_info[3])
elif len(mod_info) == 8:
scan_param = 'dihedral'
atom1 = int(mod_info[1])
atom2 = int(mod_info[2])
atom3 = int(mod_info[3])
atom4 = int(mod_info[4])
if 'Coordinates (Angstroms)' in line:
# Anchor point in log file.
xyz_flag = 1
coord_data = [ ]
if xyz_flag == 1 and '---' in line:
if dash_flag == 0:
# First dashed line after anchor and before coordinates.
dash_flag = 1
elif dash_flag == 1:
# Second dashed line after anchor and coords, turn flags off.
xyz_flag = 0
dash_flag = 0
if xyz_flag == 1 and dash_flag == 1 and '---' not in line:
coord_data = coord_data + [line]
if 'SCF Done' in line:
energy = float(line.split()[4])
if 'Optimization completed' in line:
# Only grab SCF energy for optimized structure.
scf_data = scf_data + [energy]
coord_data_LoL = coord_data_LoL + [coord_data]
# Reset geometry list.
coord_data = [ ]
counter = 1
for geom in coord_data_LoL:
if counter < 10: counter = '0' + str(counter) output_name = basename + '_step-' + str(counter) + '.xyz' # Will overwrite xyz files with this exact name. output_xyz = open(output_name,'w') output_xyz.write(str(len(geom)) + '\n') output_xyz.write(basename + '_step-' + str(counter) + '.xyz' + ' SCF Energy = ' + str(scf_data[int(counter) - 1]) + '\n') for atom in geom: atom_num = atom.split()[0] if scan_param == 'distance': if atom_num == str(atom1): mod_atom_sym1 = periodic_table[int(atom.split()[1])] if atom_num == str(atom2): mod_atom_sym2 = periodic_table[int(atom.split()[1])] elif scan_param == 'angle': if atom_num == str(atom1): mod_atom_sym1 = periodic_table[int(atom.split()[1])] if atom_num == str(atom2): mod_atom_sym2 = periodic_table[int(atom.split()[1])] if atom_num == str(atom3): mod_atom_sym3 = periodic_table[int(atom.split()[1])] elif scan_param == 'dihedral': if atom_num == str(atom1): mod_atom_sym1 = periodic_table[int(atom.split()[1])] if atom_num == str(atom2): mod_atom_sym2 = periodic_table[int(atom.split()[1])] if atom_num == str(atom3): mod_atom_sym3 = periodic_table[int(atom.split()[1])] if atom_num == str(atom4): mod_atom_sym4 = periodic_table[int(atom.split()[1])] atom_sym = periodic_table[int(atom.split()[1])] X_coord = '%10.5f' % float(atom.split()[3]) Y_coord = '%10.5f' % float(atom.split()[4]) Z_coord = '%10.5f' % float(atom.split()[5]) # Padding the xyz lines to match Gaussian input format. XYZ_string = '%-3s' '%16s' '%16s' '%16s' % (atom_sym, X_coord, Y_coord, Z_coord) # Writing optimized .xyz files. output_xyz.write(XYZ_string + '\n') counter = int(counter) + 1 # Writing output .csv file if steps have been taken. if len(scf_data) > 0:
output_csv = open(basename + '.csv','w')
output_csv.write('Scan step' + ',' + 'SCF Energy' + '\n')
for i in range(len(scf_data)):
output_csv.write(str(i + 1) + ',' + str(scf_data[i]) + '\n')
# Creating matplotlib data.
steps = range(0, len(scf_data))
plt.plot(steps, scf_data,'ro')
plt.axis([0, len(scf_data) + 1, 1.00001 * min(scf_data), 0.99999 * max(scf_data)])
# Turn offset 'off' since y axis is large numbers not changing much.
# This is needed to show y axis tick marks correctly.
plt.ticklabel_format(useOffset = False)
plt.grid(True)
plt.xlabel('Step number')
plt.ylabel('SCF energy (hartrees)')
# Sets the plot perfectly in the view window.
plt.tight_layout()
plt.show()
# Printing information to screen.
print ''
print 'Parsing through input log file: ' + input_file
if len(scf_data) == 0:
print 'No optimization steps found'
else:
print str(len(scf_data)) + ' optimizations completed.'
if scan_param == 'distance':
print 'Extracting all optimized geometries and SCF energies for the distance scan between ' + mod_atom_sym1 + str(atom1) + ' - ' + mod_atom_sym2 + str(atom2)
elif scan_param == 'angle':
print 'Extracting all optimized geometries and SCF energies for the angle scan between ' + mod_atom_sym1 + str(atom1) + ' - ' + mod_atom_sym2 + str(atom2) + ' - ' + mod_atom_sym3 + str(atom3)
elif scan_param == 'dihedral':
print 'Extracting all optimized geometries and SCF energies for the dihedral scan between ' + mod_atom_sym1 + str(atom1) + ' - ' + mod_atom_sym2 + str(atom2) + ' - ' + mod_atom_sym3 + str(atom3) + ' - ' + mod_atom_sym4 + str(atom4)
print ''
else:
print ''
print 'Please provide a Gaussian .log file from a modredundant scan as input.'
print ''