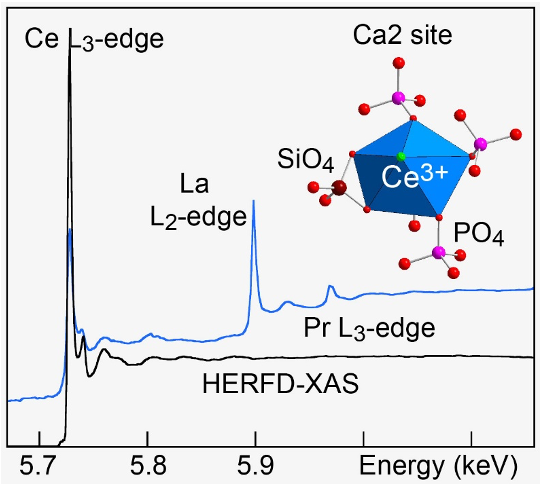
Rare earth elements can be found incorporated in apatites. Here, we studied the prototypical incorporation of Ce in fluorapatite by high-energy-resolution fluorescence-detected extended X-ray absorption fine structure (HERFD-EXAFS) spectroscopy and DFT. Stay tuned: The corresponding project, coordinated by Alain Manceau, is just starting!