")
")
Le photochromisme.
Le photochromisme consiste en un changement réversible de couleur d’un composé sous l’influence d’une irradiation généralement dans le domaine de l’UV.
Le phénomène de photochromisme peut trouver de nombreuses applications entre autre dans les domaines des verres adaptatifs (comme les lunettes qui se tentent au soleil), des mémoires optiques ou des interrupteurs moléculaires. Aujourd’hui, l’effort de développement de matériaux photochromiques se porte principalement sur des matériaux moléculaires à base de molécules organiques. Les familles des diaryléthènes (cf Figure 1) et des azobenzènes sont les plus étudiées. L’exposition aux UV de ces molécules peut conduire à la formation de liaisons chimiques ou induire des réarrangements intra-moléculaires, responsables de l’activité photochromique. L’attractivité des systèmes organiques leur vient de l’efficacité de la chimie organique à pouvoir modifier à façon les molécules afin d’adapter leur propriété aux applications souhaitées.
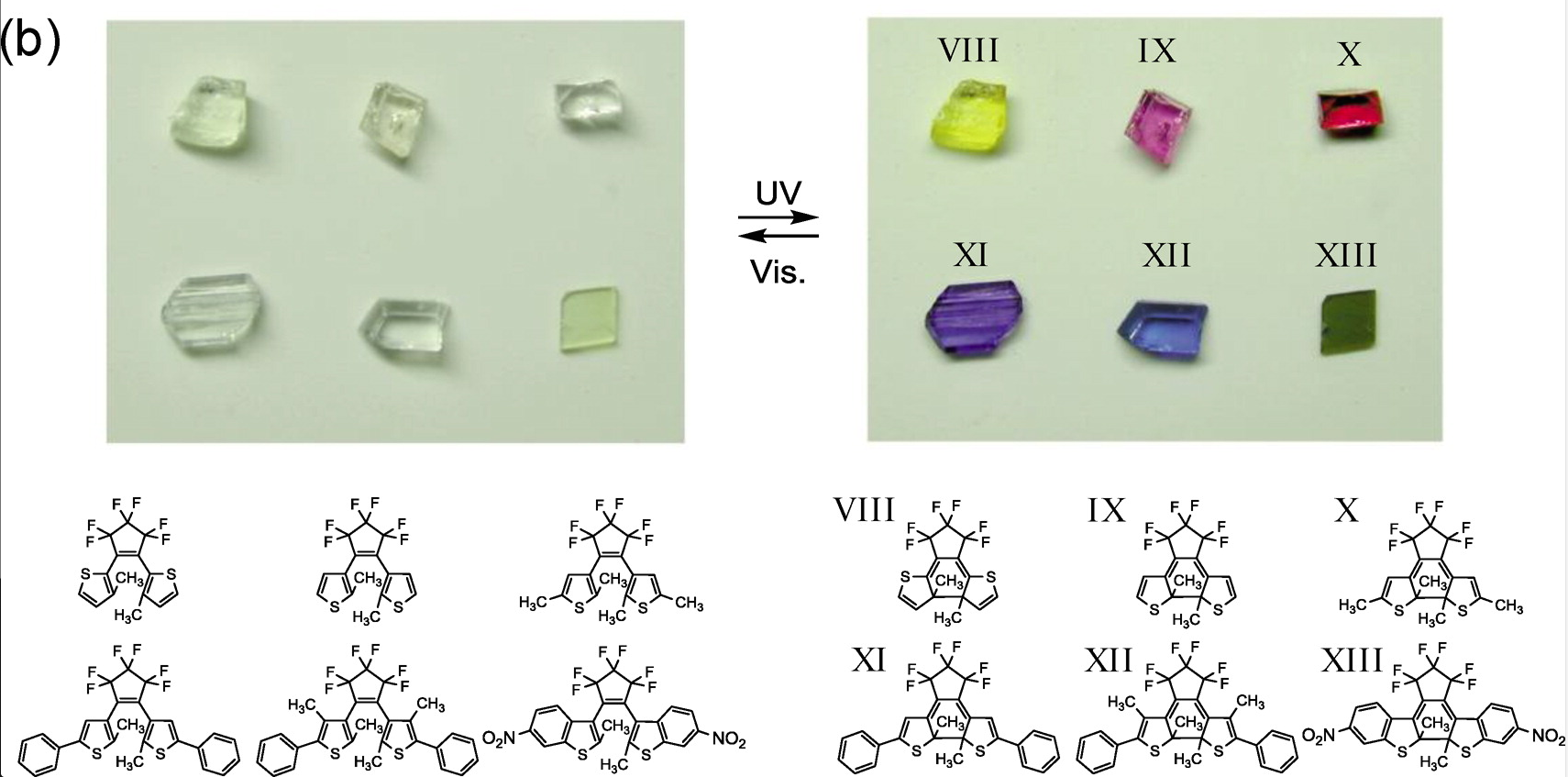
Figure 1 : matériaux moléculaires constitués de molécules de diaryléthènes. (Reprinted with permission from (Irie et al. Chem. Rev. 2014, 114, 12174). Copyright (2017) American Chemical Society)1.
Une communauté essaie néanmoins de développer des systèmes photochromiques inorganiques, typiquement des oxydes de tungstène (WO3) ou de molybdène (MoO3). L’inconvénient des matériaux inorganiques est leur moins grande adaptabilité comparée aux matériaux organiques même s’ils possèdent de nombreux avantages comme une bonne stabilité thermique et vis à vis de l’humidité.
La ténébrescence
La ténébrescence est le nom donné en géologie pour parler du phénomène de photochromisme.
Bien loin de ces recherches de nouveaux matériaux pour des applications en haute technologie se trouvent les minéraux naturels ténébrescents, uniquement connus des géologues et de quelques initiés. D’un point de vue historique, la ténébrescence des minéraux naturels est reportés depuis le début du XXième siècle.2,3
En particulier, le photochromisme de certaines sodalites, de formule Na8(SiAlO6)Cl2, a fait l’objet d’études assez approfondies depuis le milieu du XXième siècle et dans les années 70-80.4–12 La ténébrescence de ces sodalites se caractérise par un passage d’une forme incolore à une coloration violette sous l’influence d’une irradiation de 250-275 nm (4,5 - 5 eV). Très rapidement, il est apparu que le mécanisme de ce phénomène faisait intervenir des impuretés à base de soufre, probablement l’ion S22-.
Pour bien comprendre le mécanisme du photochromisme proposé dans la littérature, il faut bien avoir en tête la structure cristalline de la sodalite. La sodalite cristallise dans une structure cubique de groupe d’espace P-43n Les atomes de Si, Al et O forme une structure appelée cage-β. A l’intérieur de cette cage se trouve un tétraèdre d’ions Na+ et un ion Cl- se place au centre de ce tétraèdre (Figure 2).
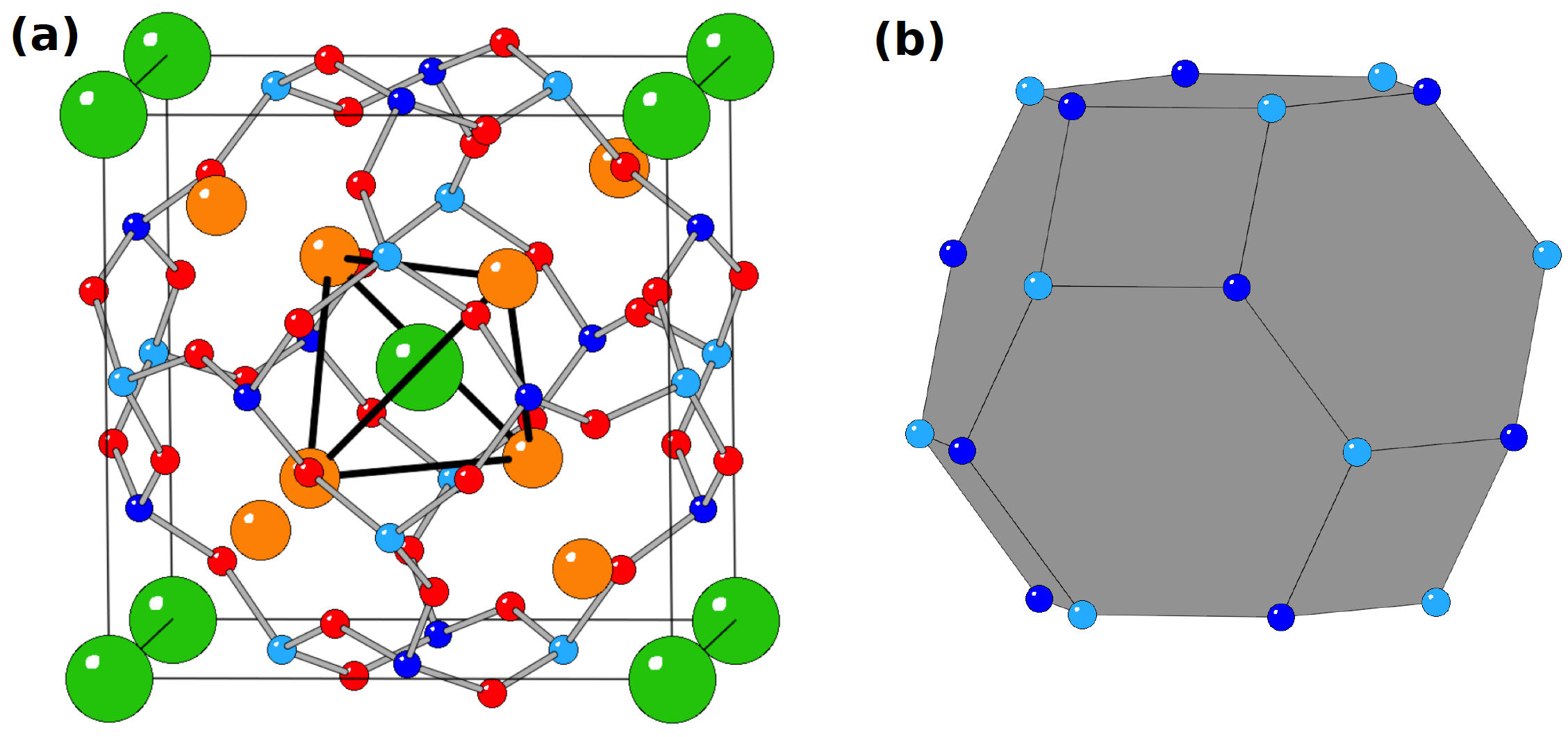
Figure 2 : (a) Représentation de la maille sodalite. Les atomes de Si, Al, Na, Cl et O sont respectivement en bleu, cyan, orange, vert et rouge. Les liaisons Si-O et Al-O sont représentées en trait gris et les côtés du tétraèdre d’ions Na+ sont représentés par des traits noirs. Reprinted with permission from (A. Curutchet and T. Le Bahers, Inorg. Chem. 2017, 56, 414-423). Copyright (2017) American Chemical Society (b) représentation schématique de la cage-β.
Le mécanisme proposé pour le photochromsime est le suivant 13 (Figure 3) :
1) (1) L’ion S22- substitue un ion Cl- dans le cristal, induisant la formation d’une lacune de Cl- pour garder l’électroneutralité du cristal.
2) (2) L’irradiation par des photons UV conduit l’ion S22- à transférer un électron dans la lacune VCl.
3) (3) Un électron piégé dans une lacune possède des niveaux d’énergie quantifiés conduisant, conduisant le matériau à absorber la lumière dans le domaine du visible.
4) (4) Sous l’influence de la lumière blanche ou de la chaleur, l’électron piégé revient vers l'ion S2-, rendant la couleur blanche au matériau.

Figure 3 : Principe du fonctionnement de la ténébrescence des sodalites. A droite, photo d’une sodalite naturelle ténébrescente (adapted from ref [17] with permission from the Royal Society of Chemistry).
Compte tenu des propriétés spectroscopiques particulières de la sodalite contenant des impuretés au soufre par rapport à la sodalite pure, on donne généralement le nom de hackmanite à cette première.
L’étude de ces minéraux est tombée en désuétudes pendant quelques décennies. Mais un regain d’intérêt s’est amorcé cette décennie porté par les travaux de M. Weller et M. Lastusaari.14–17 Ils ont montré que non seulement l’hackmanite pouvait être synthétisée en laboratoire assez facilement, mais que la composition du minéral pouvait être changée induisant des modifications de couleur de la forme colorée. Ces travaux ont ouverts la voie à une optimisation des hackmanites pour des applications comme matériaux photochromiques avec autant de versatilité que les molécules organiques.
D’un point de vue fondamental, d’autres minéraux naturels comme les scapolites et les tugtupites sont aussi connus pour leur ténébrescence.18 Mais, force est de constater, que ceux-ci ont encore été moins étudiés que les hackmanites et que le mécanisme proposé pour leur photochromisme est supposé similaire à celui de l’hackmanite.
Références
1 M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277.
2 E. W. Claffy, Am. Mineral., 1953, 38, 919.
3 S. C. Lind and D. C. Bardwell, J. Franklin Inst., 1923, 196, 375–390.
4 D. B. Medved, Am. Miner., 1954, 39, 615–629.
5 R. D. Kirk, Am. Miner., 1955, 40, 22–31.
6 H. D. Miser and J. J. Glass, Am. Mineral., 1941, 26, 437–445.
7 P. S. Pizani and M. C. Terrile, Am. Miner., 1985, 70, 1186–1192.
8 L. T. Todd, E. F. Farrell and A. Linz, IEEE Trans. Electron Devices, 1976, 23, 1183–1184.
9 V. P. Denks, A. E. Dudel’zak, C. B. Luschchik, T. V. Ruus, N. P. Soschchin and T. I. Trofimova, J. Appl. Spectrosc., 1976, 24, 23–28.
10 I. F. Chang and A. Onton, J. Electron. Mater., 1973, 2, 17–46.
11 D. W. G. Ballentyne and K. L. Bye, J. Phys. D Appl. Phys., 1970, 3, 1438–1443.
12 C. Z. Van Doorn, D. J. Schipper and P. T. Bolwijn, J. Electrochem. Soc., 1972, 119, 85–92.
13 A. Curutchet and T. Le Bahers, Inorg. Chem., 2017, 56, 414–423.
14 I. Norrbo, P. Gluchowski, I. Hyppänen, T. Laihinen, P. Laukkanen, J. Mäkelä, F. Mamedov, H. S. Santos, J. Sinkkonen, M. Tuomisto, A. Viinikanoja and M. Lastusaari, ACS Appl. Mater. Inter., 2016, 8, 11592–11602.
15 I. Norrbo, P. Gluchowski, P. Paturi, J. Sinkkonen and M. Lastusaari, Inorg. Chem., 2015, 54, 7717–7724.
16 E. R. Williams, A. Simmonds, J. A. Armstrong and M. T. Weller, J. Mater. Chem., 2010, 20, 10883–10887.
17 J. A. Armstrong and M. T. Weller, Chem. Comm., 2006, 4, 1094–1096.
18 C. C. Milisenda, S. Koch, S. Müller, T. Stephan and M. Wild, Proceedings, IGC 2015, Vilnius, 2009, 107–109.