Low-quality libraries in scRNA-seq data can arise from a variety of sources such as cell damage during dissociation or failure in library preparation. These usually manifest as “cells” with low total counts, few expressed genes and high mitochondrial or spike-in proportions. These low-quality libraries are problematic as they can contribute to misleading results in downstream analyses:
They form their own distinct cluster(s), complicating the interpretation of the results.
They distort the characterization of population heterogeneity during variance estimation or principal component analysis.
They contain genes that appear to be strongly “upregulated” due to aggressive scaling to normalize for small library sizes.
To avoid—or at least mitigate—these problems, we need to remove these cells at the start of the analysis. This step is commonly referred to as quality control (QC) on the cells.
QC metrics
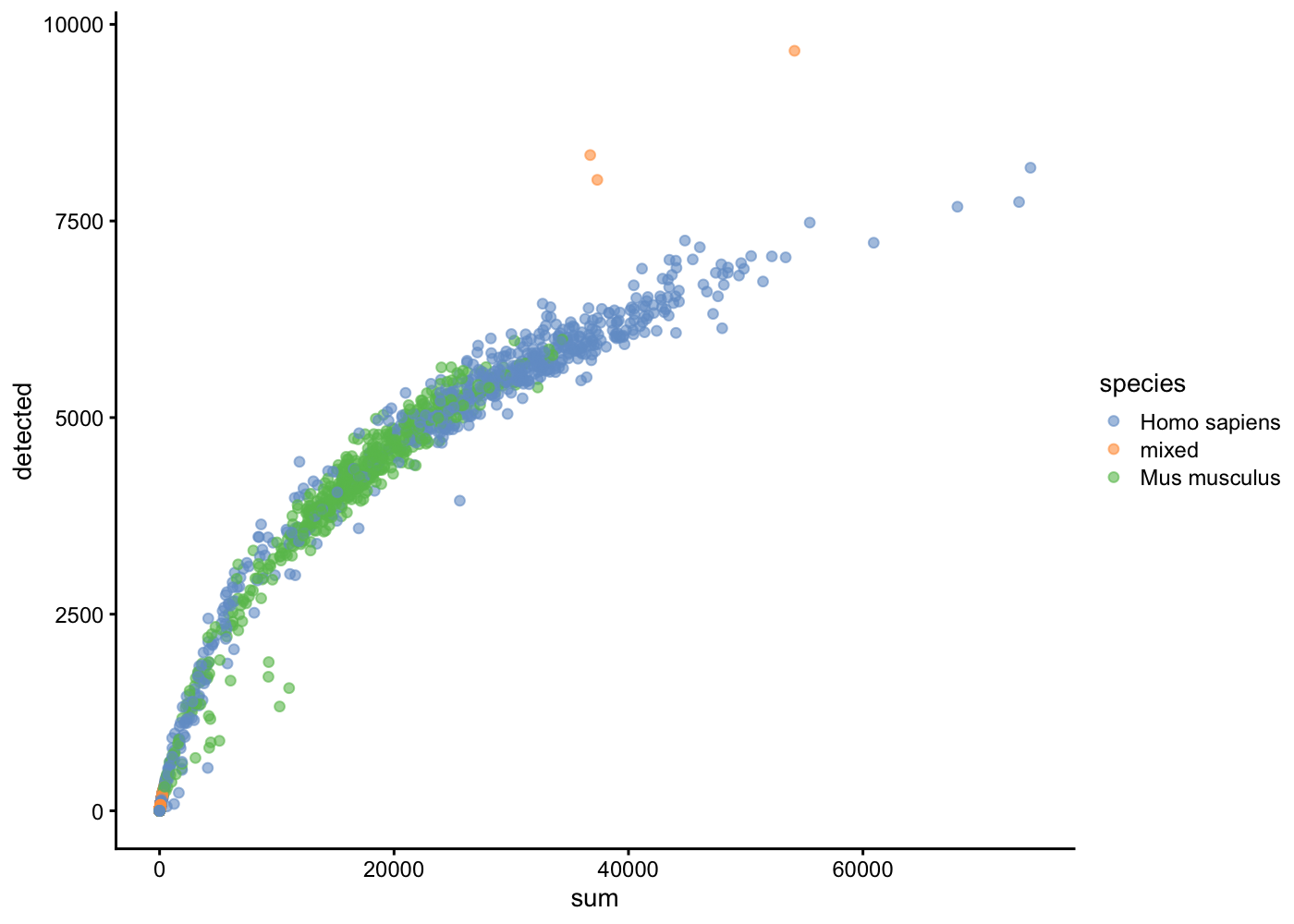
The library size is defined as the total sum of counts across all relevant features for each cell.
The number of expressed features in each cell is defined as the number of endogenous genes with non-zero counts for that cell.
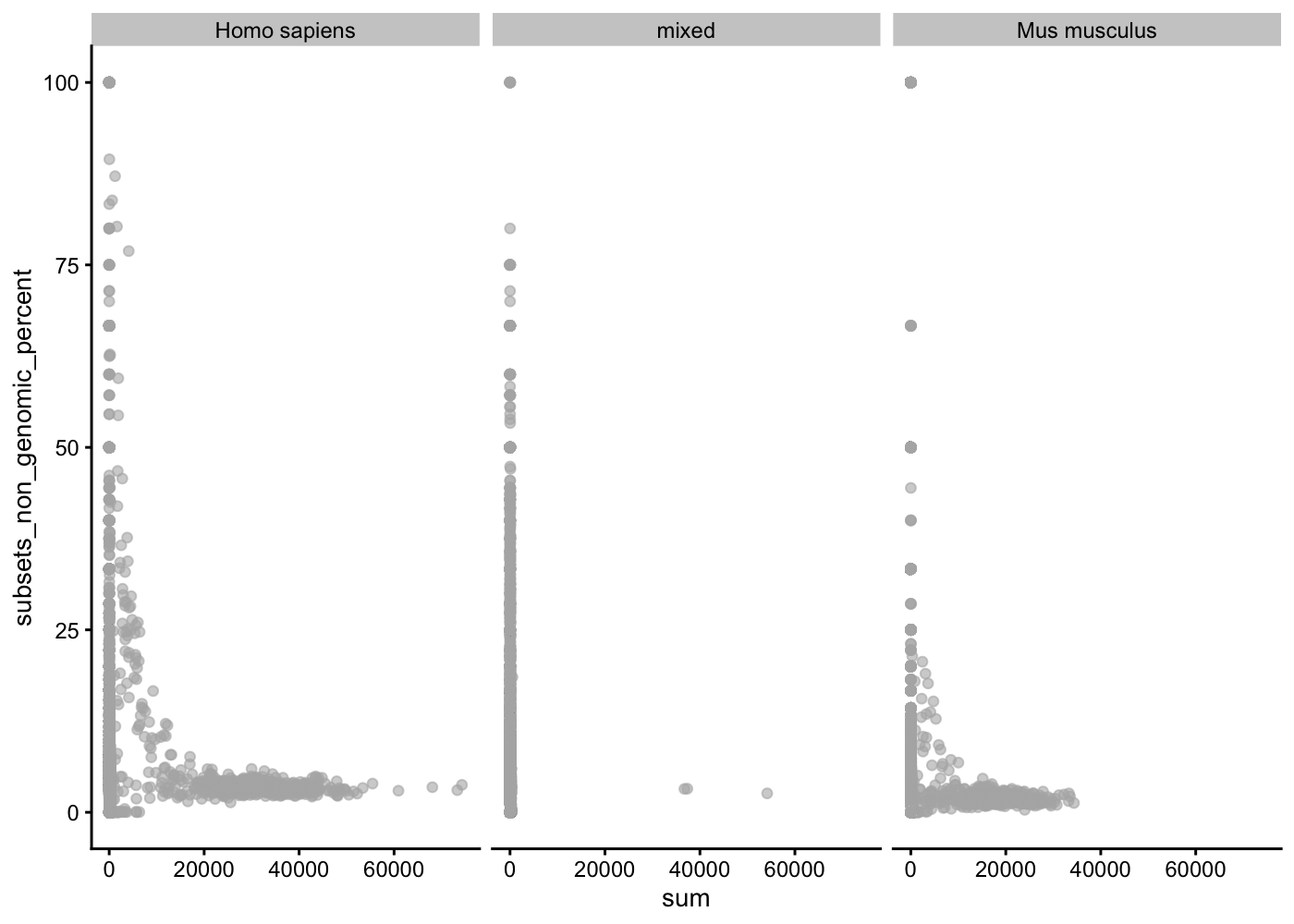
The proportion of reads mapped to spike-in transcripts is calculated relative to the total count across all features (including spike-ins) for each cell.
In the absence of spike-in transcripts, the proportion of reads mapped to genes in the mitochondrial genome can be used (and we have the chromosomic position of each gene).
A key assumption here is that the QC metrics are independent of the biological state of each cell. Poor values (e.g., low library sizes, high mitochondrial proportions) are presumed to be driven by technical factors rather than biological processes, meaning that the subsequent removal of cells will not misrepresent the biology in downstream analyses.
The first step is to remove non-expressed genes, this will reduce the dimensionality of the data we have to work with. To test if a given gene is expressed you can use the nexprs() function and add the information to the rowData() slot.
We can compute a large number of different quality metrics on each gene and on each cell. The scater package contains the addPerCellQC() function that appends a lot of quality control data. The subset option allows us to compute statistics on the non-genomic gene content (i.e. Mitocondrial genes)
We can identify outlier cells. The isOutlier function provides a more data-adaptive way of choosing these thresholds. This defines the threshold at a certain number of median absolute deviations (MADs) away from the median. Values beyond this threshold are considered outliers and can be filtered out, assuming that they correspond to low-quality cells. Here, we define small outliers (using type="lower") for the log-total counts at 3 MADs from the median. Use this function to identify cells with low total umi counts.
colData(sce)$keep_total <- scater::isOutlier(colData(sce)$sum, type ="lower", log =TRUE)
Analogous to our call to addPerCellQC() in the prior section, the addPerFeatureQC() function will insert values in the rowData slot of our sce object:
sce <- scater::addPerFeatureQC(sce)rowData(sce)
DataFrame with 77202 rows and 8 columns
genes_id genes_names species
<character> <character> <character>
ENSG00000237235.2 ENSG00000237235.2 NA Homo sapiens
ENSG00000223997.1 ENSG00000223997.1 NA Homo sapiens
ENSG00000228985.1 ENSG00000228985.1 NA Homo sapiens
ENSG00000282253.1 ENSG00000282253.1 NA Homo sapiens
ENSG00000282431.1 ENSG00000282431.1 NA Homo sapiens
... ... ... ...
ENSMUSG00000106908.1 ENSMUSG00000106908.1 NA Mus musculus
ENSMUSG00000107077.1 ENSMUSG00000107077.1 NA Mus musculus
ENSMUSG00000106901.1 ENSMUSG00000106901.1 NA Mus musculus
ENSMUSG00000107196.1 ENSMUSG00000107196.1 NA Mus musculus
ENSMUSG00000118095.1 ENSMUSG00000118095.1 NA Mus musculus
chr_pos is_genomic expressed mean detected
<character> <logical> <logical> <numeric> <numeric>
ENSG00000237235.2 14 TRUE FALSE 0 0
ENSG00000223997.1 14 TRUE FALSE 0 0
ENSG00000228985.1 14 TRUE FALSE 0 0
ENSG00000282253.1 CHR_HSCHR7_2_CTG6 FALSE FALSE 0 0
ENSG00000282431.1 7 TRUE FALSE 0 0
... ... ... ... ... ...
ENSMUSG00000106908.1 CHR_MG3562_PATCH FALSE FALSE 0 0
ENSMUSG00000107077.1 CHR_MG3562_PATCH FALSE FALSE 0 0
ENSMUSG00000106901.1 CHR_MG3562_PATCH FALSE FALSE 0 0
ENSMUSG00000107196.1 CHR_MG3562_PATCH FALSE FALSE 0 0
ENSMUSG00000118095.1 CHR_MG4249_PATCH FALSE FALSE 0 0
summary(rowData(sce)$detected)
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00000 0.00000 0.00000 0.03016 0.01436 6.48303
Filter empty droplets
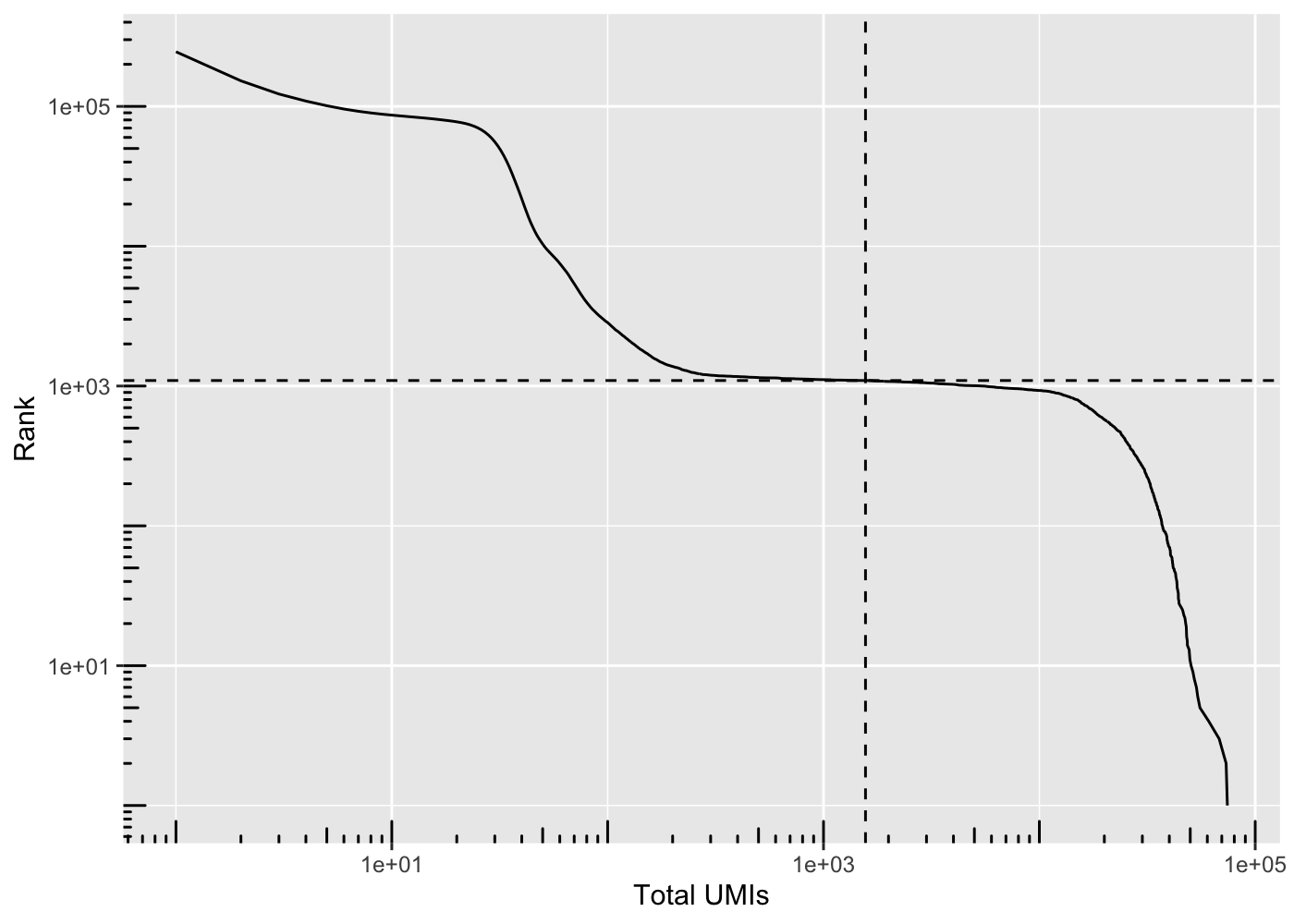
Most barcodes in the matrix correspond to empty droplets. A common way to determine which barcodes are empty droplets and which are real cells is to plot the rank of total UMI counts of each barcode against the total UMI counts itself, which is commonly called knee plot. The inflection point in that plot, signifying a change in state, is used as a cutoff for total UMI counts; barcodes below that cutoff are deemed empty droplets and removed.
Droplet-based data is that we have no prior knowledge about whether a particular library (i.e., cell barcode) corresponds to cell-containing or empty droplets. Thus, we need to call cells from empty droplets based on the observed expression profiles.
The distribution of total counts exhibits a sharp transition between barcodes with large and small total counts, probably corresponding to cell-containing and empty droplets respectively. A simple approach would be to apply a threshold on the total count to only retain those barcodes with large totals. However, this unnecessarily discards libraries derived from cell types with low RNA content.
To compute the barcode ranks, you can use the barcodesRanks() function. The following knee_plot function allows you to display the knee plot.
We can also use the emptyDrops() function to test whether the expression profile for each cell barcode is significantly different from the ambient RNA pool (Lun et al. 2019). Any significant deviation indicates that the barcode corresponds to a cell-containing droplet.
# emptyDrops performs Monte Carlo simulations to compute p-values,# so we need to set the seed to obtain reproducible results.set.seed(100)ed_out <- DropletUtils::emptyDrops(counts(sce))table(Sig=ed_out$FDR <=0.05, Limited=ed_out$Limited)is_cell <- ed_out$FDR <=0.05sum(is_cell, na.rm=TRUE)tibble(logprob = ed_out$LogProb,is_cell = is_cell,total = ed_out$Total,species =colData(sce)$species) %>%ggplot() +geom_point(aes(x = total, y =-logprob, color = is_cell)) +annotation_logticks() +scale_y_log10() +scale_x_log10() +facet_wrap(~species) +labs(x ="Total UMI count", y ="-Log Probability")colData(sce)$is_cell <-ifelse(is.na(is_cell), FALSE, is_cell)
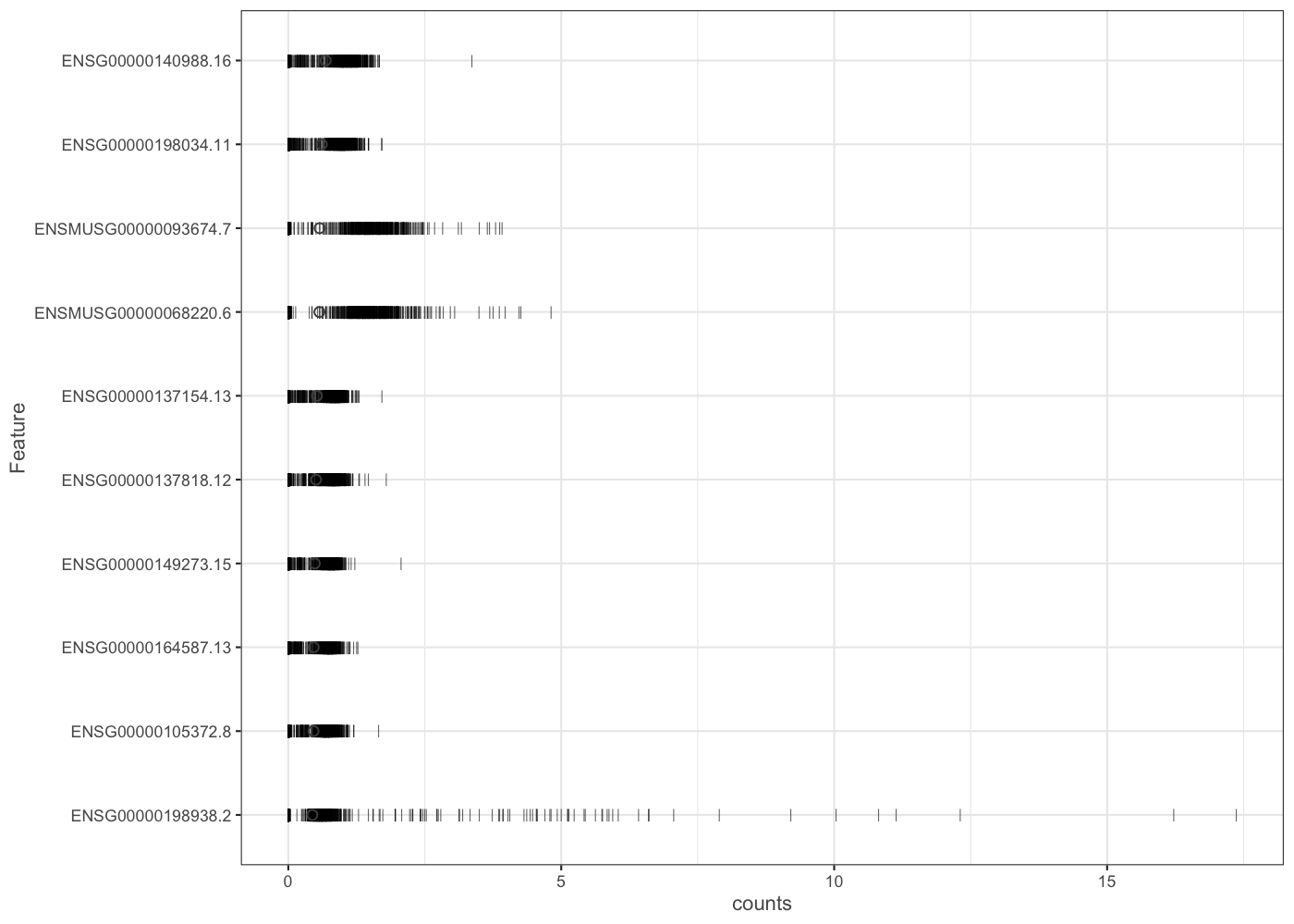
Now that we can work with a more reasonable number of cells (with the is_cell variable), we can plot genes. The plotHighestExprs() fuction shows the top 50 (by default) most-expressed genes Each row in the plot below corresponds to a gene, and each bar corresponds to the expression of a gene in a single cell. The circle indicates the median expression of each gene, with which genes are sorted.
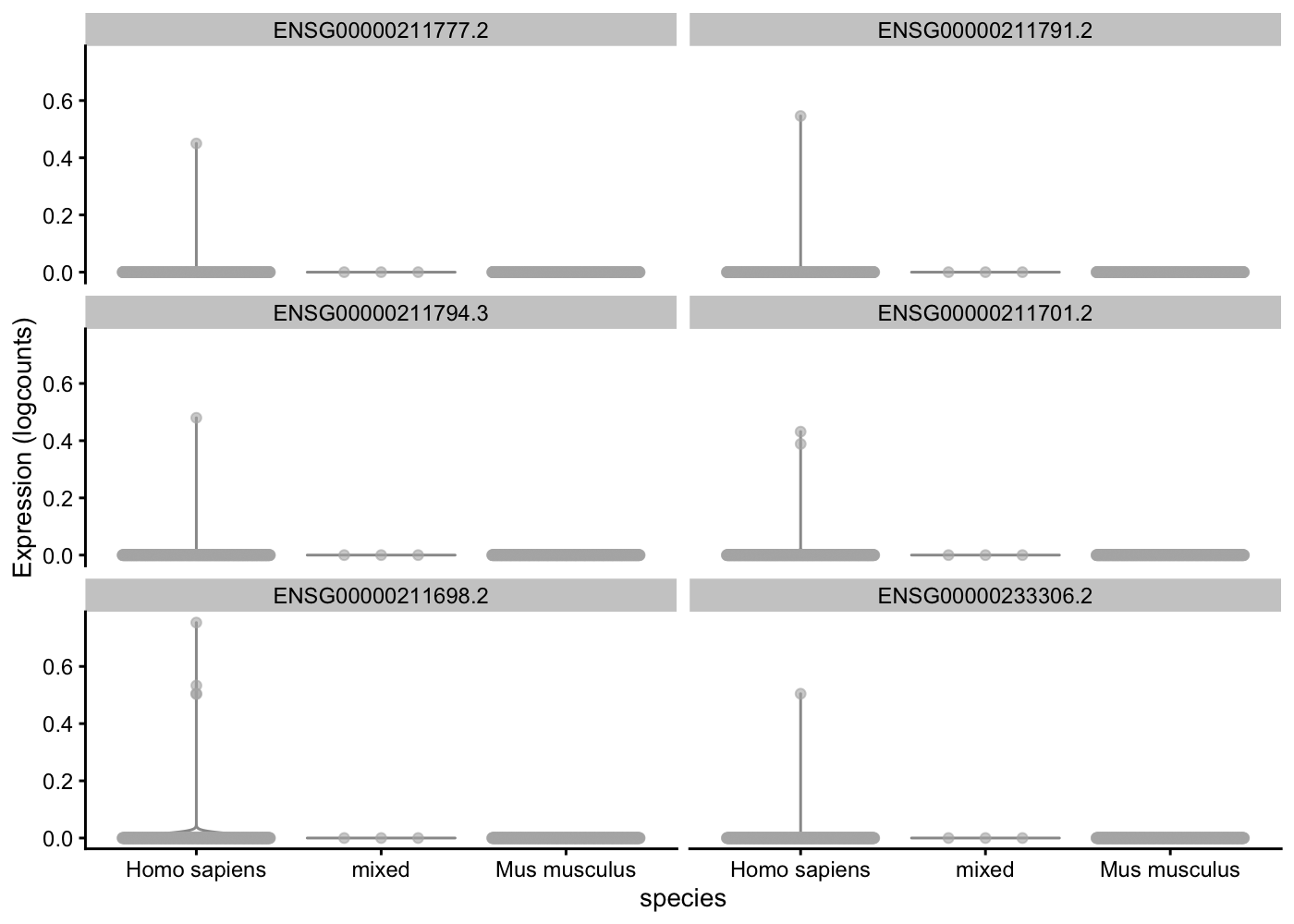
You can plot a selection of genes with the plotExpression() function (the 6 firsts).
Here, we added a logNormCounts matrix to our sce object. We can store many different counts matrix simultaneously in one sce object. This is very useful to switch between function working on counts or on pseudo-count. We see that the assay slot has grown to contain two entries: "counts" (our initial data) and "logcounts" (the log-transformed normalized data). Similar to "counts", the "logcounts" name can be conveniently accessed using logcounts(sce).
logcounts(sce)
Doublet detection
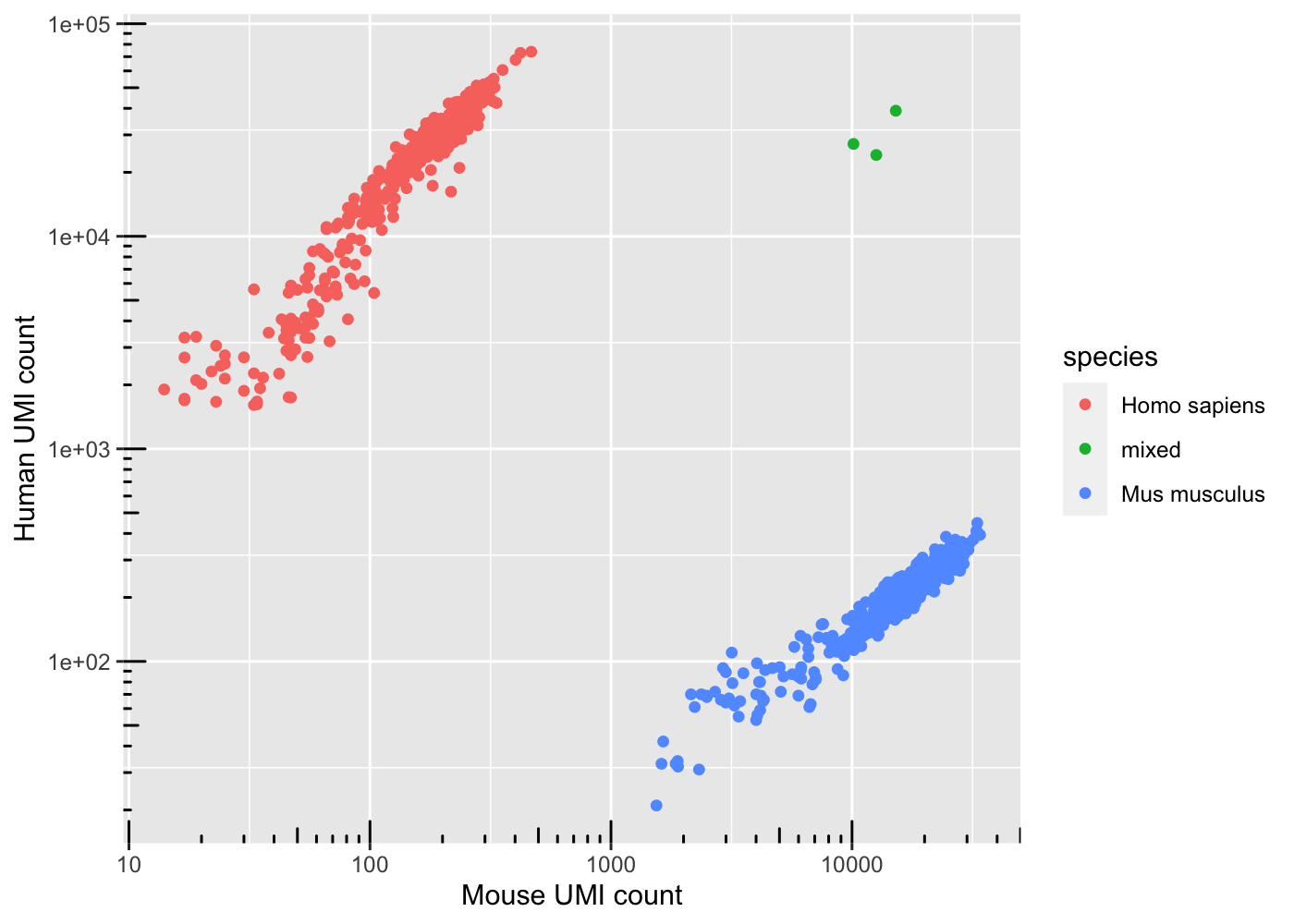
We have seen many cells annotated as mixed, how many real cells are human-mouse mixture ? Make a visualization of the proportion of n_mm_umi and n_hg_umi for each cells.
colData(sce) %>%as_tibble() %>% dplyr::filter(is_cell) %>%ggplot() +geom_point(aes(x = n_mm_umi, y = n_hg_umi, color = species)) +annotation_logticks() +scale_y_log10() +scale_x_log10() +labs(x ="Mouse UMI count", y ="Human UMI count")
How many cells are from humans and how many from mice? The number of cells with mixed species indicates doublet rate. Here, it’s easy, we can identify human-mouse mixed droplets and deduce that they are doublet. But what about human-human doublets or mouse-mouse doublets ?
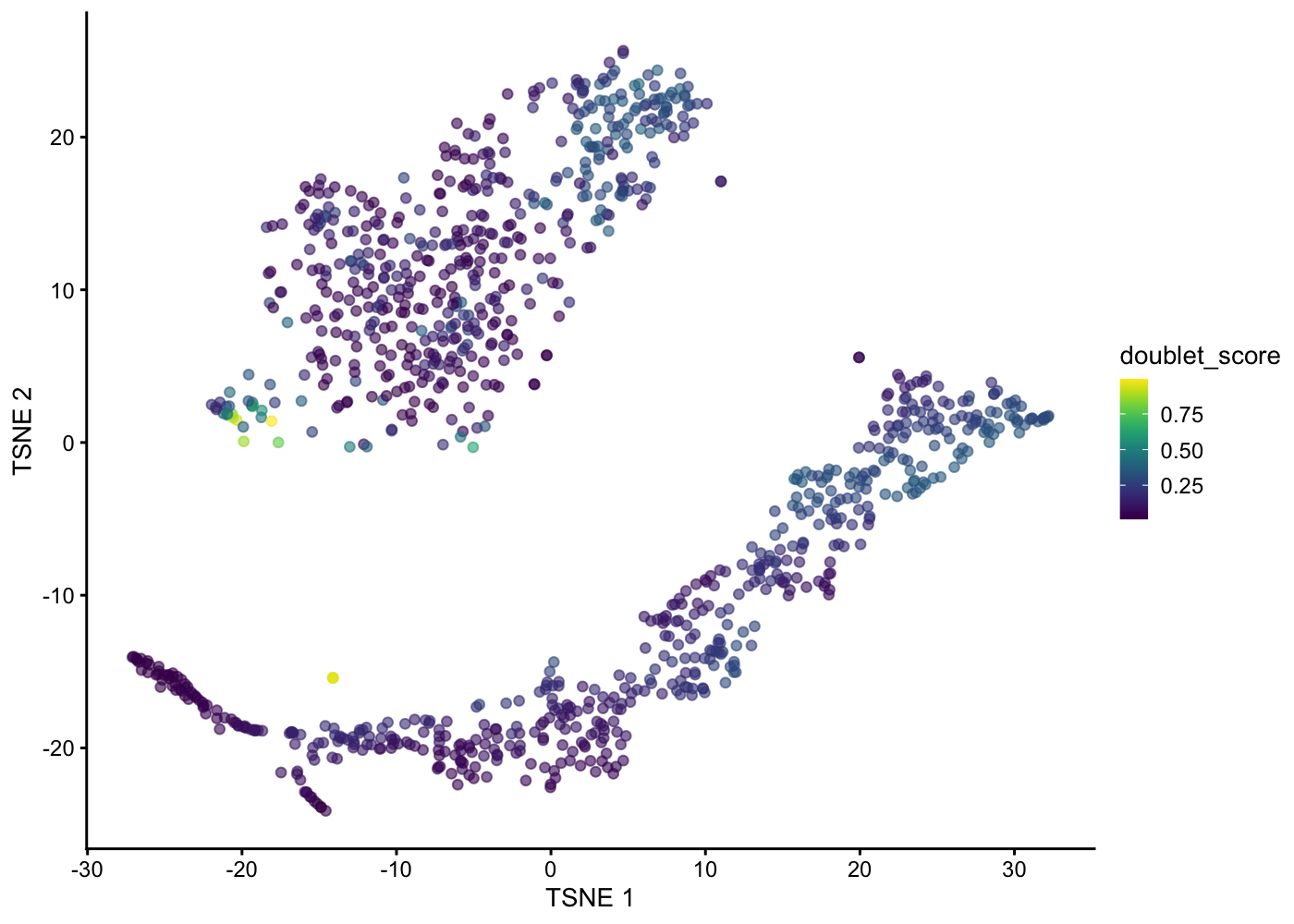
Doublet detection is a difficult problem, and state-of-the-art strategy involves in silico simulation of doublets from the single-cell expression profiles (Dahlin et al. 2018). This is performed using the doubletCells() function from scran, which will :
Simulate thousands of doublets by adding together two randomly chosen single-cell profiles.
For each original cell, compute the density of simulated doublets in the surrounding neighborhood.
For each original cell, compute the density of other observed cells in the neighborhood.
Return the ratio between the two density as a “doublet score” for each cell.
This approach assumes that the simulated doublets are good approximations for real doublets. The use of random selection accounts for the relative abundances of different subpopulations, which affect the likelihood of their involvement in doublets; and the calculation of a ratio avoids high scores for non-doublet cells in highly abundant subpopulations.
Doublet detection procedures should only be applied to libraries generated in the same experimental batch. It is obviously impossible for doublets to form between two cells that were captured separately.